DoS resolution in conduction bands, and prtdos
Posted: Sun Mar 25, 2018 6:47 am
I'm doing a GaN (Gallium Nitride) calculation in wurtzite crystal structure (4 atoms: 2 Ga, 2 N), and I'm having issues generating a good quality DoS plot, as well as learning about the relevant abinit details.
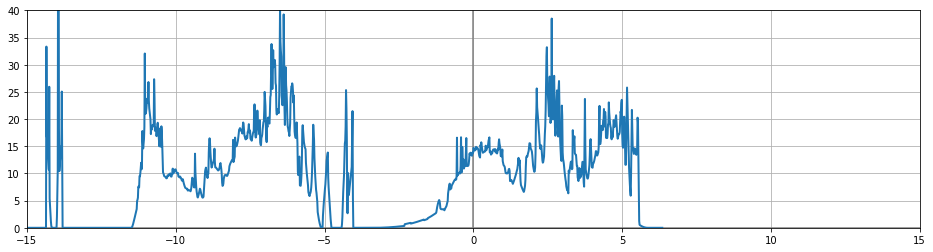
After going through the tutorials, I thought there's no discussion on generating a good quality DoS, so it must be easy to extract one from the main calculation (the scf run after all convergence studies, and before the band-structure run). This was further supported by the fact that an abipy example plotted dos from the GSR.nc file from the main run. I did a similar thing with my calculation but the plotted dos is of low resolution / bad quality. The input file and dos plot are attached:
So I started looking, and it turns out there are a host of DoS generating variables (prtdos, dosdeltae, pawprtdos) that are not discussed at all in the first four tutorials, and barely touched upon in the PAW tutorial. prtdos documentation is not bad, but still doesn't help with the pros and cons of the various options. Plus it says prtdos 1 requires convergence study, but fails to mention what kind.
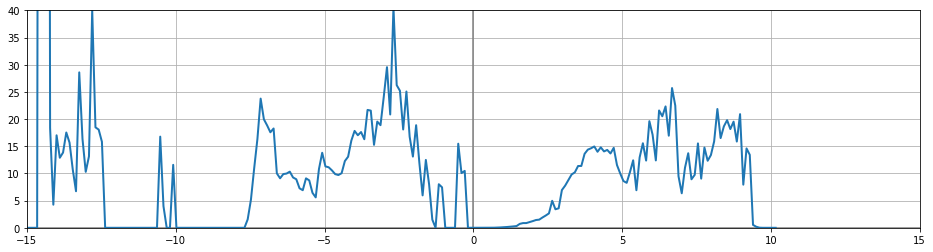
I did a prtdos 1 run as well and the result does not look much better. I tried prtdos 2, the resolution looks better for the valence band (it has sharper features) but conduction band looks bad, as given below:
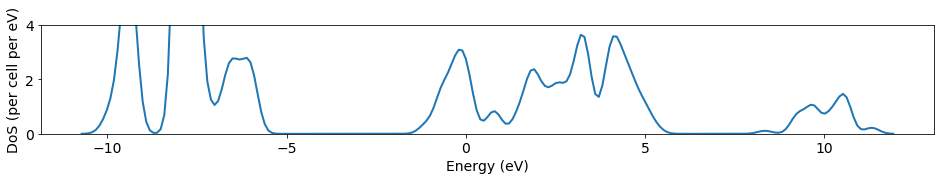
Here, by conduction band I mean the bump between 0.3 and 0.45 Ha. This bump should look different, for reference the DoS for GaN (from Diakite et al, Calculated Electronic and Related Properties of Wurtzite and Zinc Blende Gallium Nitride (GaN), 2014) looks like this:
So I thought maybe I need to specify a nband value like in a band-structure calculation (I had to use nband 16 to get conduction bands), so I used nband 16 with prtdos 2 and now I'm getting the error:
This error is against the extra parameters added to the input file shown above, as follows:
The DEN file comes from the main calculation (which was done after k point and ecut convergence, as well as structural relaxation of acell as well as atomic positions. The relaxed structure was manually copied into the input file shown above). The log file of that main calculation mentions nband of 19, so I'm not sure why abinit is complaining about nband 16.
Kindly advise as to what should be the next course of action. Thank you.
Also since I'm using PAW dataset, should I use prtdos 3. I still haven't read that section of prtdos and it looks a bit complicated but I can try.
Finally regarding nband 19 in the main calculation, which I did not specify, what if I want to do the main calculation (i.e., the generation of the DEN file) with higher number of bands? (btw, am I correct in thinking that the key output of the main abinit calculation is the DEN file, and once we have the DEN file we don't need anything else for band-structure and DoS and all other information, unless for efficiency reasons, e.g., we might use WFK to speed things up, I don't know.)
After going through the tutorials, I thought there's no discussion on generating a good quality DoS, so it must be easy to extract one from the main calculation (the scf run after all convergence studies, and before the band-structure run). This was further supported by the fact that an abipy example plotted dos from the GSR.nc file from the main run. I did a similar thing with my calculation but the plotted dos is of low resolution / bad quality. The input file and dos plot are attached:
Code: Select all
kptopt 1
nshiftk 1
shiftk 0.0 0.0 0.5
ngkpt 6 6 2
toldfe 1.0e-10
acell 6.0911781167E+00 6.0911781167E+00 9.9164297984E+00 Bohr
rprim 1.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 8.6602540378E-01 1.6076496088E-36
6.1232339957E-17 1.0605752387E-16 1.0000000000E+00
natom 4
ntypat 2
typat 1 2 1 2
znucl 31.00000 7.00000
xred 6.6666666667E-01 3.3333333333E-01 3.7593954042E-01
6.6666666667E-01 3.3333333333E-01 -9.3954042085E-04
3.3333333333E-01 6.6666666667E-01 8.7593954042E-01
3.3333333333E-01 6.6666666667E-01 4.9906045958E-01
ecut 25.0
ecutsm 0.5
pawecutdg 50
nstep 50
diemac 12.0
ixc -101130
- gsrdos.png (21.98 KiB) Viewed 7812 times
So I started looking, and it turns out there are a host of DoS generating variables (prtdos, dosdeltae, pawprtdos) that are not discussed at all in the first four tutorials, and barely touched upon in the PAW tutorial. prtdos documentation is not bad, but still doesn't help with the pros and cons of the various options. Plus it says prtdos 1 requires convergence study, but fails to mention what kind.
I did a prtdos 1 run as well and the result does not look much better. I tried prtdos 2, the resolution looks better for the valence band (it has sharper features) but conduction band looks bad, as given below:
- prtdos2.png (19.35 KiB) Viewed 7812 times
Here, by conduction band I mean the bump between 0.3 and 0.45 Ha. This bump should look different, for reference the DoS for GaN (from Diakite et al, Calculated Electronic and Related Properties of Wurtzite and Zinc Blende Gallium Nitride (GaN), 2014) looks like this:
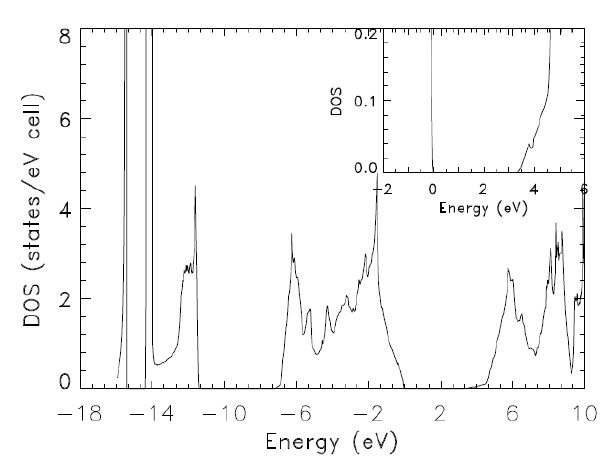
- refwgandos.png (28.32 KiB) Viewed 7812 times
So I thought maybe I need to specify a nband value like in a band-structure calculation (I had to use nband 16 to get conduction bands), so I used nband 16 with prtdos 2 and now I'm getting the error:
Code: Select all
--- !ERROR
src_file: m_dtset.F90
src_line: 278
mpi_rank: 0
message: |
Initialization of occ, with occopt= 1
There are not enough bands to get charge balance right
Action: modify input file ...
(check the pseudopotential charges, the variable charge,
and the declared number of bands, nband)
This error is against the extra parameters added to the input file shown above, as follows:
Code: Select all
nshiftk 2
shiftk
0.0 0.0 0.0
0.0 0.0 0.5
ngkpt 8 8 4
nband 16
prtdos 2
iscf -3
irdden 1
The DEN file comes from the main calculation (which was done after k point and ecut convergence, as well as structural relaxation of acell as well as atomic positions. The relaxed structure was manually copied into the input file shown above). The log file of that main calculation mentions nband of 19, so I'm not sure why abinit is complaining about nband 16.
Kindly advise as to what should be the next course of action. Thank you.
Also since I'm using PAW dataset, should I use prtdos 3. I still haven't read that section of prtdos and it looks a bit complicated but I can try.
Finally regarding nband 19 in the main calculation, which I did not specify, what if I want to do the main calculation (i.e., the generation of the DEN file) with higher number of bands? (btw, am I correct in thinking that the key output of the main abinit calculation is the DEN file, and once we have the DEN file we don't need anything else for band-structure and DoS and all other information, unless for efficiency reasons, e.g., we might use WFK to speed things up, I don't know.)