Hai Abinit Users
I would like to know whether the "Fundamental gap" obtained in the log file after band structure computation, will give the exact band gap between the valence and conduction band.
Can I take the "Fundamental gap" value as the band gap value.
Any comment on this are highly appreciated.
Thanks and Regards
Seba
Regarding band gap calculation
Moderator: bguster
Re: Regarding band gap calculation
If by exact you mean within the capabilities of the LDA, then yes. If you mean the experimental single-particle band gap, then no. In principle, the difference between the lowest unoccupied and the highest occupied state plus a derivative discontinuity in the exchange-correlation energy would give you the exact band gap. In practice, none of the currently available approximations of the xc-potential are able to reproduce the discontinuity. Sometimes it is taken from experimental data, in which case it is called the scissor operator. Also, note that the fundamental gap as reported by Abinit in general depends on Brillouin zone sampling.
Raul Laasner
Netherlands Institute for Space Research
Netherlands Institute for Space Research
-
Seba Darshan
- Posts: 63
- Joined: Mon Feb 17, 2014 5:19 am
Re: Regarding band gap calculation
raul_l wrote:If by exact you mean within the capabilities of the LDA, then yes. If you mean the experimental single-particle band gap, then no. In principle, the difference between the lowest unoccupied and the highest occupied state plus a derivative discontinuity in the exchange-correlation energy would give you the exact band gap. In practice, none of the currently available approximations of the xc-potential are able to reproduce the discontinuity. Sometimes it is taken from experimental data, in which case it is called the scissor operator. Also, note that the fundamental gap as reported by Abinit in general depends on Brillouin zone sampling.
Dear Mr. Raul
Thank you for your reply.
I had this doubt because the values of bandgap (the gap between the valence and conduction band) from the band structure plot computed using GGA withTrouiller-Martins pseudopotentials is different from the "Fundamental gap" available under "Gap Info" in the output log file, in some cases that I studied. I am not sure of which value I can take as band gap.
Please guide me in this regard.
Thanks and Regards
Seba
Re: Regarding band gap calculation
For a converged calculation, they should be the same. Otherwise, it depends on the input file. How much do they differ in your case? Under Gap info, you can see the coordinates of the k-points that Abinit is considering as band extrema:
If these coincide with the true band extrema of your system, then the reported fundamental gap should be correct. Otherwise, you might need to increase or shift your k-mesh. Alternatively, you can extract the band gap from the calculated band structure if the k-path passes the correct band extrema. If you wish, we can investigate your input file to find the source of discrepancy.
Code: Select all
Top of valence bands at :
Bottom of conduction at :If these coincide with the true band extrema of your system, then the reported fundamental gap should be correct. Otherwise, you might need to increase or shift your k-mesh. Alternatively, you can extract the band gap from the calculated band structure if the k-path passes the correct band extrema. If you wish, we can investigate your input file to find the source of discrepancy.
Raul Laasner
Netherlands Institute for Space Research
Netherlands Institute for Space Research
-
Seba Darshan
- Posts: 63
- Joined: Mon Feb 17, 2014 5:19 am
Re: Regarding band gap calculation
raul_l wrote:For a converged calculation, they should be the same. Otherwise, it depends on the input file. How much do they differ in your case? Under Gap info, you can see the coordinates of the k-points that Abinit is considering as band extrema:Code: Select all
Top of valence bands at :
Bottom of conduction at :
If these coincide with the true band extrema of your system, then the reported fundamental gap should be correct. Otherwise, you might need to increase or shift your k-mesh. Alternatively, you can extract the band gap from the calculated band structure if the k-path passes the correct band extrema. If you wish, we can investigate your input file to find the source of discrepancy.
Dear Sir
As per your comment, I am attaching the input file that I used for calculating the band structure of a 2by2 graphene supercell with two boron heteroatoms:
acell 9.7633499650E+00 9.7633499650E+00 1.8897160000E+01 Bohr
rprim 0.86602540378 -0.5 0.0
0.86602540378 0.5 0.0
0.0 0.0 1.0
chkprim 0
ntypat 2
znucl 5 6
natom 8
typat 2 1 2 2 2 2 1 2
xred 1.5818669120E-01 1.5818669120E-01 7.3814456430E-37
3.3333333333E-01 3.3333333333E-01 7.3814456430E-37
1.5818669120E-01 6.8362661760E-01 7.3814456430E-37
3.1637338240E-01 8.4181330880E-01 7.3814456430E-37
6.8362661760E-01 1.5818669120E-01 7.3814456430E-37
8.4181330880E-01 3.1637338240E-01 7.3814456430E-37
6.6666666667E-01 6.6666666667E-01 7.3814456430E-37
8.4181330880E-01 8.4181330880E-01 -5.1670119501E-36
ecut 15
nstep 60
ndtset 2
kptopt1 1
nshiftk1 1
shiftk1 0.0 0.0 0.0
ngkpt1 6 6 1
prtden1 1
toldff1 5.0d-6
iscf2 -2
getden2 -1
kptopt2 -3
nband2 40
ndivk2 20 20 20
kptbounds2 0.0 0.0 0.0
1/3 2/3 0.0
1/2 1/2 0.0
0.0 0.0 0.0
tolwfr2 1.0d-12
enunit2 1
The relaxed acell and xred values are used in the input file. The band structure obtained is attached along with this. From the plot attached, what I understand is that bandgap is zero, at the point B in the band structure (K-point of the Briolluin zone) . The "Gap info" in the log file is as follows:
=== Gap info ===
>>>> For spin 1
Minimum optical gap = 1.5544 [eV], located at k-point : 0.1667 0.0000 0.0000
Fundamental gap = 0.6696 [eV], Top of valence bands at : 0.0000 0.0000 0.0000
Bottom of conduction at : 0.5000 0.0000 0.0000
In the gap information, they have selected the top of valence band at 0, 0, 0 and bottom of conduction band at 0.5, 0, 0 and thus calculated the band gap. I would like to know out of this which one is correct. I would like to know whether my understanding is wrong.
I am experiencing the same problem for other simulations too, so it would be of great help if you could correct me.
I would greatly appreciate if you could help me to analyse the band gap from the band structure plot attached.
Hope to hear from you favourably,
Thanks and Regards
Seba
- Attachments
-
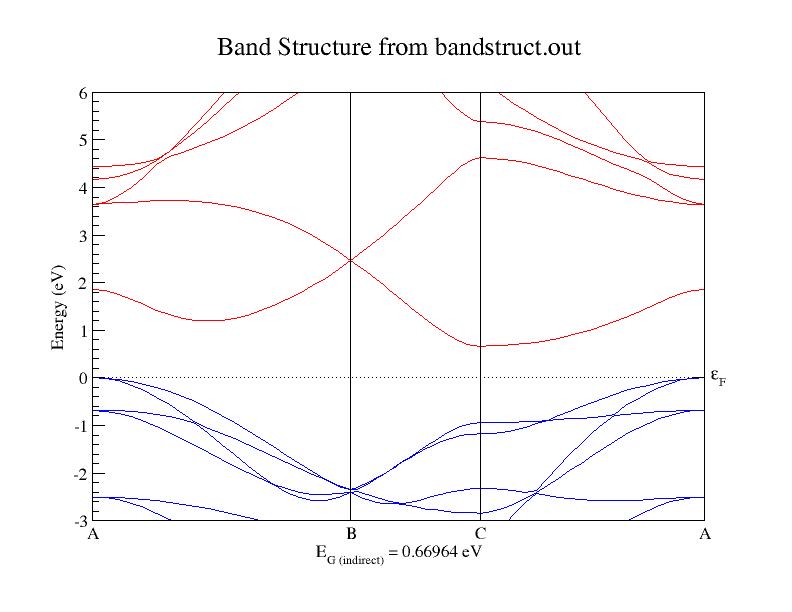
- Band structure plot for the above input file
- Band structure plot.jpg (49.26 KiB) Viewed 5690 times
Re: Regarding band gap calculation
I'm not an expert on graphene but your calculation seems to be correct. The indirect gap obtained from the band structure plot coincides with the one under gap info, so there's no problem with that. I'm not sure where you get the gap value as zero (perhaps the degeneracy at B confuses somehow). The highest occupied state at B, judging from the plot, is at around -2.2 eV. The first allowed direct transition (no momentum transfer) is to the lowest unoccupied state at B, which is about 2.5 eV. The first indirect transition (simultaneous absorption of a photon and a phonon) would be to the lowest empty state at C, which is at 0.67 eV. So B does not correspond to the minimum direct or indirect gap.
In your case, the values obtained from the band structure plot and as reported by the calculation coincide. In general, if a non-optimal k-mesh is chosen or the k-path for plotting the band structure does not pass the correct k-points, neither value would be correct.
In your case, the values obtained from the band structure plot and as reported by the calculation coincide. In general, if a non-optimal k-mesh is chosen or the k-path for plotting the band structure does not pass the correct k-points, neither value would be correct.
Raul Laasner
Netherlands Institute for Space Research
Netherlands Institute for Space Research