Note
Go to the end to download the full example code.
Wavefunction file
This example shows how to analyze the wavefunctions stored in the WFK.nc file.
================================= File Info =================================
Name: si_nscf_WFK.nc
Directory: /home/runner/work/abipy/abipy/abipy/data/refs/si_ebands
Size: 389.84 kB
Access Time: Tue Jun 16 09:08:58 2026
Modification Time: Tue Jun 16 09:01:46 2026
Change Time: Tue Jun 16 09:01:46 2026
================================= Structure =================================
Full Formula (Si2)
Reduced Formula: Si
abc : 3.866975 3.866975 3.866975
angles: 60.000000 60.000000 60.000000
pbc : True True True
Sites (2)
# SP a b c
--- ---- ---- ---- ----
0 Si 0 0 0
1 Si 0.25 0.25 0.25
Abinit Spacegroup: spgid: 0, num_spatial_symmetries: 48, has_timerev: True, symmorphic: True
============================== Electronic Bands ==============================
Number of electrons: 8.0, Fermi level: 5.598 (eV)
nsppol: 1, nkpt: 14, mband: 8, nspinor: 1, nspden: 1
smearing scheme: none (occopt 1), tsmear_eV: 0.272, tsmear Kelvin: 3157.7
Direct gap:
Energy: 2.532 (eV)
Initial state: spin: 0, kpt: $\Gamma$ [+0.000, +0.000, +0.000], band: 3, eig: 5.598, occ: 2.000
Final state: spin: 0, kpt: $\Gamma$ [+0.000, +0.000, +0.000], band: 4, eig: 8.130, occ: 0.000
Fundamental gap:
Energy: 0.524 (eV)
Initial state: spin: 0, kpt: $\Gamma$ [+0.000, +0.000, +0.000], band: 3, eig: 5.598, occ: 2.000
Final state: spin: 0, kpt: [+0.000, +0.429, +0.429], band: 4, eig: 6.123, occ: 0.000
Bandwidth: 11.856 (eV)
Valence maximum located at kpt index 6:
spin: 0, kpt: $\Gamma$ [+0.000, +0.000, +0.000], band: 3, eig: 5.598, occ: 2.000
Conduction minimum located at kpt index 12:
spin: 0, kpt: [+0.000, +0.429, +0.429], band: 4, eig: 6.123, occ: 0.000
TIP: Use `--verbose` to print k-point coordinates with more digits
PWWaveFunction: nspinor: 1, spin: 0, band: 0
<class 'abipy.core.gsphere.GSphere'>: kpoint: L [+0.500, +0.000, +0.000], ecut: 6.000000, npw: 180, istwfk: 1
Mesh3D: nx=18, ny=18, nz=18
import abipy.data as abidata
from abipy.abilab import abiopen
# Open the DEN.nc file
ncfile = abiopen(abidata.ref_file("si_nscf_WFK.nc"))
print(ncfile)
# The DEN file has a `Structure` an `ElectronBands` object and wavefunctions.
# print(ncfile.structure)
# To plot the KS eigenvalues.
# ncfile.ebands.plot()
# Extract the wavefunction for the first spin, the first band and k=[0.5, 0, 0]
wave = ncfile.get_wave(spin=0, kpoint=[0.5, 0, 0], band=0)
print(wave)
# This is equivalent to
# wave = ncfile.get_wave(spin=0, kpoint=0, band=0)
# because [0.5, 0, 0] is the first k-point in the WFK file
# To visualize the total charge wih vesta
# visu = wave.visualize_ur2("vesta"); visu()
# To plot the wavefunction along the line connecting
# the first and the second in the structure:
# wave.plot_line(point1=0, point2=1)
# wave.plot_line(point1=0, point2=1, with_krphase=True)
# alternatively, one can define the line in terms of two points
# in fractional coordinates:
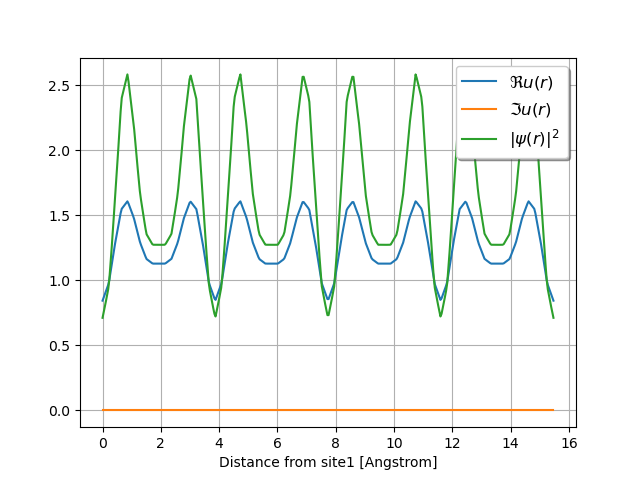
wave.plot_line(point1=[0, 0, 0], point2=[0, 4, 0], with_krphase=False, num=400)
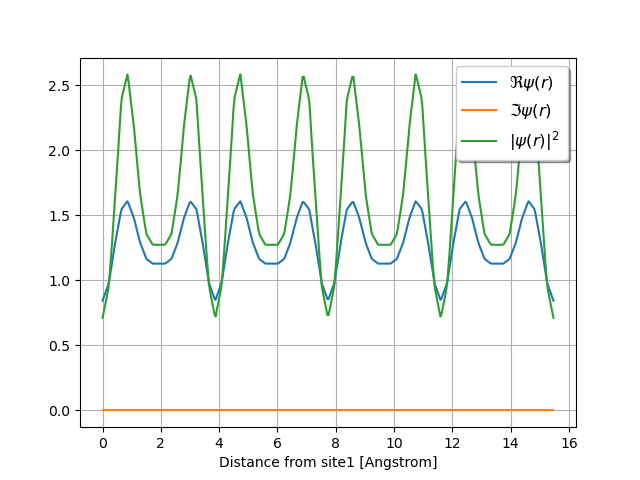
wave.plot_line(point1=[0, 0, 0], point2=[0, 4, 0], with_krphase=True, num=400)
# To plot the wavefunction along the lines connect the firt atom in the structure
# and all the neighbors within a sphere of radius 3 Angstrom:
# wave.plot_line_neighbors(site_index=0, radius=3)
ncfile.close()
Total running time of the script: (0 minutes 0.450 seconds)